File:Phylogenetic tree of coronaviruses.jpg
Ir a la navegación
Ir a la búsqueda
No disponible a mayor resolución.
Phylogenetic_tree_of_coronaviruses.jpg (571 × 451 píxeles; tamaño de archivo: 75 kB; tipo MIME: image/jpeg)
Leyendas
Leyendas
Añade una explicación corta acerca de lo que representa este archivo
Coronavirus
Resumen
[editar]{kind=link}
| Descripción |
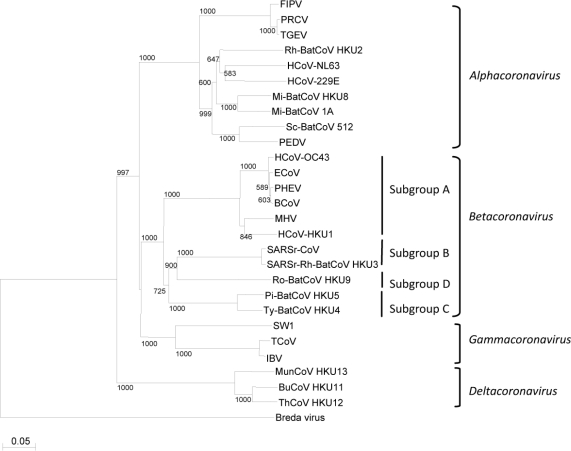
English: Phylogenetic analysis of RNA-dependent RNA polymerases (Pol) of coronaviruses with complete genome sequences available. The tree was constructed by the neighbor-joining method and rooted using Breda virus polyprotein. Bootstrap values were calculated from 1000 trees. 1118 amino acid positions in Pol were included. The scale bar indicates the estimated number of substitutions per 20 amino acids. All abbreviations for the coronaviruses were the same as those in Figure 1. |
| Fecha | |
| Fuente | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185738/ |
| Autor | Patrick C. Y. Woo, Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen |
Licencia
[editar]{kind=link}
Este archivo se encuentra bajo la licencia Creative Commons Atribución 3.0 Unported.
- Eres libre:
- de compartir – de copiar, distribuir y transmitir el trabajo
- de remezclar – de adaptar el trabajo
- Bajo las siguientes condiciones:
- atribución – Debes otorgar el crédito correspondiente, proporcionar un enlace a la licencia e indicar si realizaste algún cambio. Puedes hacerlo de cualquier manera razonable pero no de manera que sugiera que el licenciante te respalda a ti o al uso que hagas del trabajo.
Historial del archivo
Haz clic sobre una fecha y hora para ver el archivo tal como apareció en ese momento.
| Fecha y hora | Miniatura | Dimensiones | Usuario | Comentario | |
|---|---|---|---|---|---|
| actual | 02:42 7 mar 2020 | | 571 × 451 (75 kB) | Guest2625 (discusión | contribs.) | Uploaded a work by Patrick C. Y. Woo, Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185738/ with UploadWizard |
No puedes sobrescribir este archivo.
Usos del archivo
No hay páginas que enlacen a este archivo.
Uso global del archivo
Las wikis siguientes utilizan este archivo:
- Uso en bn.wikipedia.org
- Uso en en.wikipedia.org
- Uso en fa.wikipedia.org
- Uso en fr.wikipedia.org
- Uso en ig.wikipedia.org
- Uso en sq.wikipedia.org
- Uso en su.wikipedia.org
{kind=link}