File:Phylogenetic tree of coronaviruses.jpg
Aller à la navigation
Aller à la recherche
Pas de plus haute résolution disponible.
Phylogenetic_tree_of_coronaviruses.jpg (571 × 451 pixels, taille du fichier : 75 kio, type MIME : image/jpeg)
Légendes
Légendes
Ajoutez en une ligne la description de ce que représente ce fichier
Coronavirus
Description
[modifier]{kind=link}
| Description |
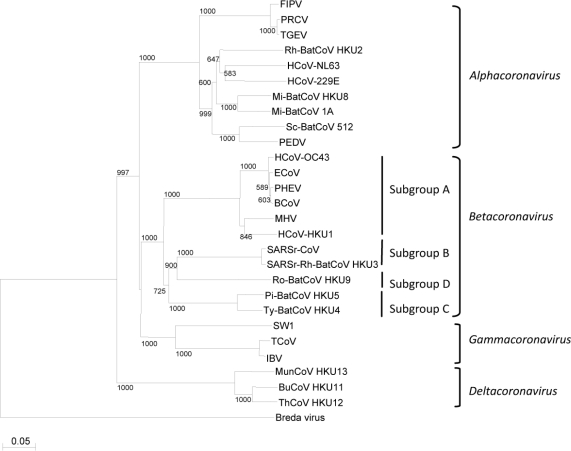
English: Phylogenetic analysis of RNA-dependent RNA polymerases (Pol) of coronaviruses with complete genome sequences available. The tree was constructed by the neighbor-joining method and rooted using Breda virus polyprotein. Bootstrap values were calculated from 1000 trees. 1118 amino acid positions in Pol were included. The scale bar indicates the estimated number of substitutions per 20 amino acids. All abbreviations for the coronaviruses were the same as those in Figure 1. |
| Date | |
| Source | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185738/ |
| Auteur | Patrick C. Y. Woo, Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen |
Conditions d’utilisation
[modifier]{kind=link}
Ce fichier est disponible selon les termes de la licence Creative Commons Attribution 3.0 Non transposée.
- Vous êtes libre :
- de partager – de copier, distribuer et transmettre cette œuvre
- d’adapter – de modifier cette œuvre
- Sous les conditions suivantes :
- paternité – Vous devez donner les informations appropriées concernant l'auteur, fournir un lien vers la licence et indiquer si des modifications ont été faites. Vous pouvez faire cela par tout moyen raisonnable, mais en aucune façon suggérant que l’auteur vous soutient ou approuve l’utilisation que vous en faites.
Historique du fichier
Cliquer sur une date et heure pour voir le fichier tel qu'il était à ce moment-là.
| Date et heure | Vignette | Dimensions | Utilisateur | Commentaire | |
|---|---|---|---|---|---|
| actuel | 7 mars 2020 à 02:42 | | 571 × 451 (75 kio) | Guest2625 (d | contributions) | Uploaded a work by Patrick C. Y. Woo, Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185738/ with UploadWizard |
Vous ne pouvez pas remplacer ce fichier.
Utilisations locales du fichier
Aucune page n’utilise ce fichier.
Utilisations du fichier sur d’autres wikis
Les autres wikis suivants utilisent ce fichier :
- Utilisation sur bn.wikipedia.org
- Utilisation sur en.wikipedia.org
- Utilisation sur fa.wikipedia.org
- Utilisation sur fr.wikipedia.org
- Utilisation sur ig.wikipedia.org
- Utilisation sur sq.wikipedia.org
- Utilisation sur su.wikipedia.org
{kind=link}